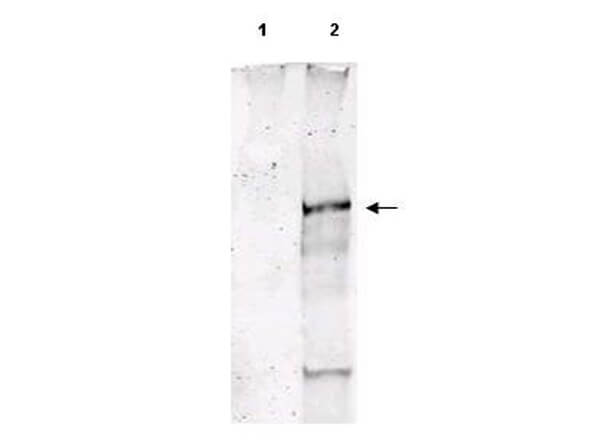
ATM phospho S1981 Biotin Conjugated Antibody
Mouse Monoclonal 10H11.E12 IgG1 kappa
$50.00 to US & $70.00 to Canada for most products. Final costs are calculated at checkout.
Background
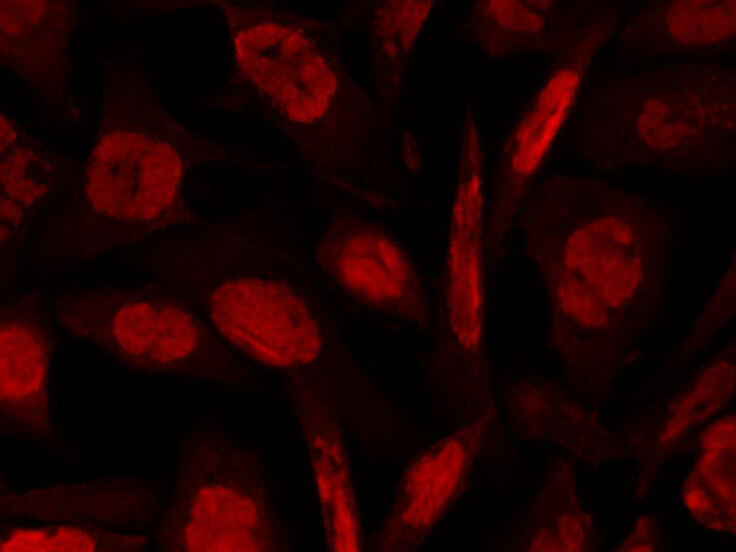
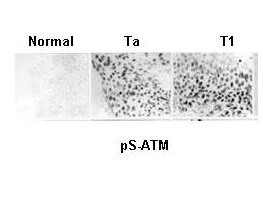
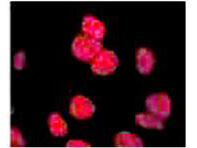
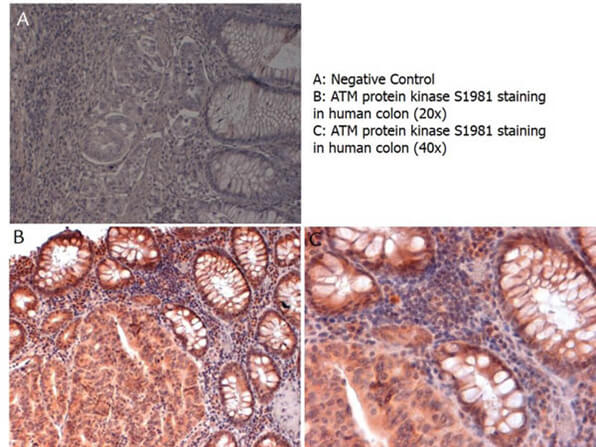
ATM, the gene mutated in the hereditary disease ataxia-telangiectasia, codes for a protein kinase that acts as a master regulator of cellular responses to DNA double-strand breaks. ATM is normally inactive and the question of how it is activated in the event of DNA damage (due to ionizing radiation for instance) is central to understanding its function. ATM protein is now shown to be present in undamaged cells as an inactive dimer. Low doses of ionizing radiation, which induce only a few DNA breaks, activate at least half of the total ATM protein present, possibly in response to changes in chromatin structure. The ATM gene encodes a 370-kDa protein that belongs to the phosphoinositide 3-kinase (PI(3)K) superfamily, but which phosphorylates proteins rather than lipids. The 350-amino-acid kinase domain at the carboxy terminus of this large protein is the only segment of ATM with an assigned function. Exposure of cells to IR triggers ATM kinase activity, and this function is required for arrests in G1, S and G2 phases of the cell cycle. Several substrates of the ATM kinase participate in these IR-induced cell-cycle arrests. These include p53, Mdm2 and Chk2 in the G1 checkpoint; Nbs1, Brca1, FancD2 and SMC1 in the transient IR-induced S-phase arrest; and Brca1 and hRad17 in the G2/M checkpoint. Ideal for Cancer, Cell Signaling, Chromatin, Neuroscience and Signal Transduction research.
Product Details
Target Details
Application Details
Formulation
Shipping & Handling
This product is for research use only and is not intended for therapeutic or diagnostic applications. Please contact a technical service representative for more information. All products of animal origin manufactured by Rockland Immunochemicals are derived from starting materials of North American origin. Collection was performed in United States Department of Agriculture (USDA) inspected facilities and all materials have been inspected and certified to be free of disease and suitable for exportation. All properties listed are typical characteristics and are not specifications. All suggestions and data are offered in good faith but without guarantee as conditions and methods of use of our products are beyond our control. All claims must be made within 30 days following the date of delivery. The prospective user must determine the suitability of our materials before adopting them on a commercial scale. Suggested uses of our products are not recommendations to use our products in violation of any patent or as a license under any patent of Rockland Immunochemicals, Inc. If you require a commercial license to use this material and do not have one, then return this material, unopened to: Rockland Inc., P.O. BOX 5199, Limerick, Pennsylvania, USA.